基本配置做好,接下来就是将各种各样的数据整合进circos环图里了,本章主要介绍links、highlights、text数据作图。
Links
Links主要试图利用连线关系,表明染色体上不同位置之间的联系。这在实际生物学问题中很常用,因为基因组往往在功能调控、空间结构上存在相互作用关系,而网络图就是最佳的展示方式之一。
我们可以利用<<include>>单独引入links配置文件,也可以在主配置文件circos.conf中添加<links>模块来实现这一功能:
show_links = yes
<links>
# 调用show_links的值
show = conf(show_links)
# Circos提供两种link方式,一种是bezier适合点对点连线,一种是ribbon适合片对片链接
ribbon = yes
# ribbon的画法是start1->end1->end2->start2,故可能出现twist,设置flat = yes取消twist
# 同样地,可以设置twist = yes强行制造twist
flat = yes
# links的起始终止位置
radius = 0.45r-55p
# Circos提供bezier_radius、crest、bezier_radius_purity等参数来控制link的弯曲形式,具体见下图
# 若希望设置为undef,建议在<link>子模块中设置
bezier_radius = 0r
color = black_a5
# 对于ribbon,还可以通过stroke_color和stroke_thickness来设置边缘
<link>
# 指定数据文件位置
file = data/links.txt
# 这里不额外讲rules的使用方法,举些例子供参考吧
<rules>
<rule>
# 只有link数据才存在chr、size等前缀+1、2的情况
# 表示当size1(link起始结点)的大小小于1Mb时
condition = var(size1) < 1e6
# 符合上述条件的link线不显示
show = no
</rule>
# 注意这里,Circos中是从前至后依次进行rule判断的,若满足第一个rule,则不继续判断后续rule了
# 可通过flow参数改变这一规则
<rule>
# 这里相当于总为true
condition = 1
# eval表示赋值
# 这里即把chr2(link末端结点)的染色体名称去掉前两个字母后,组合进"chr%s_a3",再赋给color
color = eval(sprintf("chr%s_a3",substr(var(chr2),2)))
# remap表示取值范围映射,这里即把size2(link末端结点)的值由[0,10e6]映射至[0,-100],然后赋给z
z = eval(remap_int(var(size2),0,10e6,0,-100))
</rule>
</rules>
</link>
</links>
这里看一下官网提供的关于曲线弯曲设置的示意图:
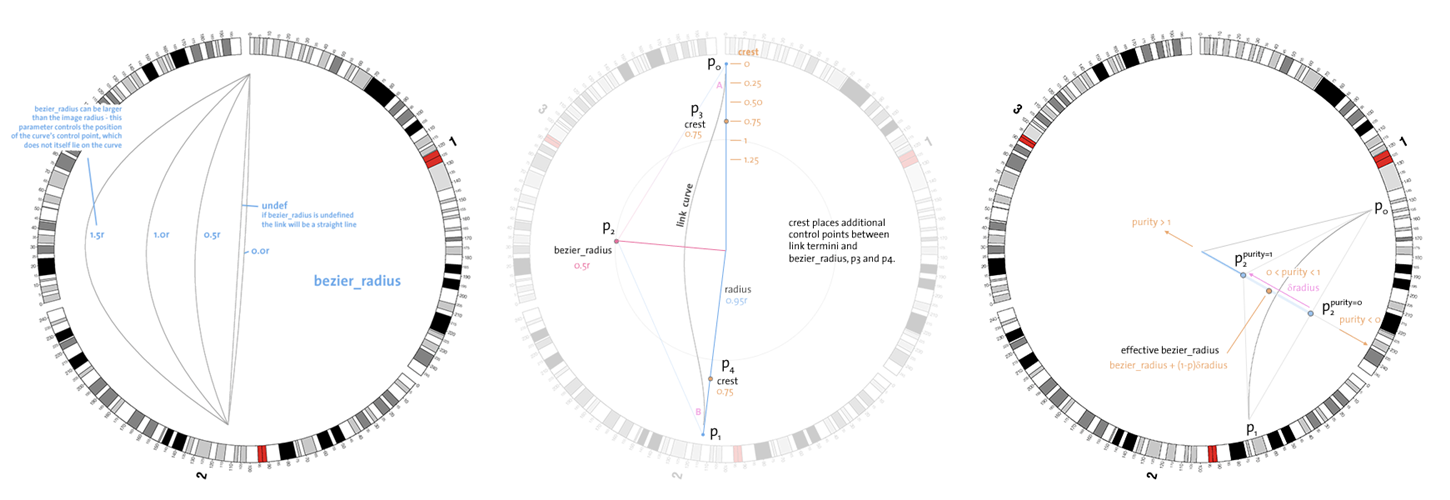 Circos很有意思地提供了perturb等参数,它们在指定范围内随机取值,从而可以让不同
Circos很有意思地提供了perturb等参数,它们在指定范围内随机取值,从而可以让不同<link>子模块随机呈现不同曲线效果:
# 设置是否使用perturb
perturb = yes
# 设为0表示不使用perturb
perturb_crest = 0
# new_value = value * [ pmin + (pmax-pmin)*urd ],其中urd在[0,1)间平均分布
perturb_bezier_radius = 0.5,1.2
perturb_bezier_radius_purity = 0.5,1
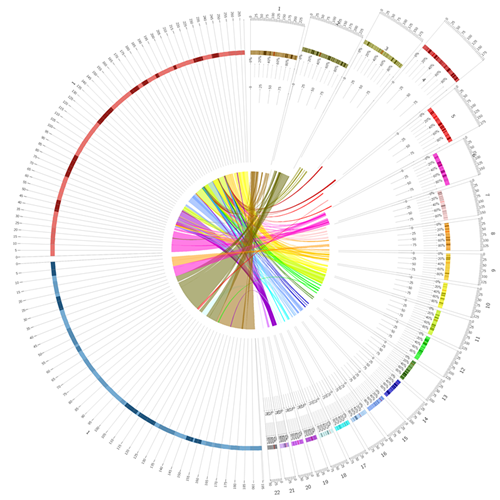
接下来,我们看一下links模块的展示效果:
Highlights
高亮显示是一个很常用的功能,在Circos中,你既可以用它来标注感兴趣的不同区域,也可以仅仅用来装饰整个环图。Highlight包括两种使用方式,一种是利用<highlights>模块,再一种是设置type = highlight的<plot>子模块。从图形展示的角度来说区别不大,唯一就是<highlights>模块所呈现的数据在其它数据层的下面,<plot>模块则反之。不过<highlights>模块还有个方便之处,是可以更好地设置在染色体上显示(利用ideogram = yes,而不需设置r0和r1,从而显示得更完美)。
Highlight的基本数据格式只需要染色体名称和起始终止位置即可,也可以通过补充设置fill_color、r0、r1等覆盖默认值,达到特殊显示的需要。下面,我们设置两个type = highlight的<plot>子模块作为例子:
show_highlights = yes
<plots>
# highlight1
<plot>
show = conf(show_highlights)
# 表示这是一个highlight类型的plot
type = highlight
# 指定数据文件位置
file = data/highlights.txt
# highlight区块的内外半径,实际不严格区分哪个在内哪个在外
r0 = 1r+180p
r1 = 1r+181p
# 边缘的颜色、粗细
stroke_color = black
stroke_thickness = 1
# 填充色
fill_color = red
</plot>
# highlight2
<plot>
show = conf(show_highlights)
type = highlight
file = data/highlights.txt
r0 = 0.45r-50p
r1 = 0.45r-45p
# 通过rules,设置颜色和karyotype保持一致
<rules>
<rule>
# Circos使用Perl语法规则,这里即正则表达式
# 设置所有人类染色体的颜色
condition = var(chr) =~ /hs/
fill_color = eval(lc sprintf("chr%s",substr(var(chr),2)))
</rule>
# 设置小鼠染色体的颜色
<rule>
condition = var(chr) =~ /mm/
fill_color = blues-5-seq-4
</rule>
# 设置大鼠染色体的颜色
<rule>
condition = var(chr) =~ /rn/
fill_color = reds-5-seq-4
</rule>
</rules>
</plot>
</plots>
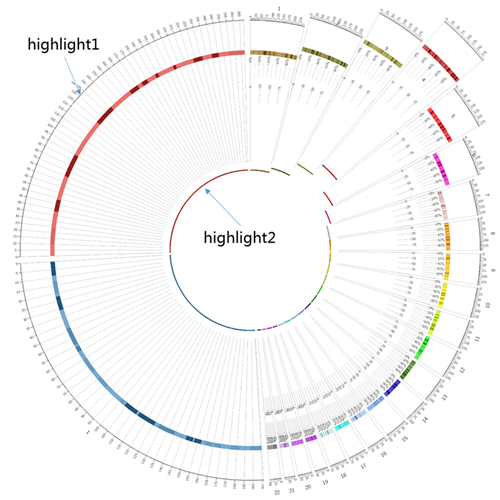
下面是展示效果,可以看到,这里就仅仅利用highlight模块作为装饰:
Text
基因组数据的分析和作图少不了对一些区域进行注释,所以text的显示是很重要的内容。Text是一个type = text的<plot>子模块,其基本数据格式除了要求染色体名称和起始终止位置外,还应有value一栏,即text的内容。下面例子使用了人类基因组中部分omim和cancer相关基因名称的数据。值得注意的是,通常在大量数据情况下,text不能完整显示,特别是邻近位置,所以对于text排版的设置尤为重要:
show_text = yes
<plots>
<plot>
show = conf(show_text)
# 表示这是一个text类型的plot
type = text
file = data/text.txt
color = black
# 通过r0和r1来设定显示半径,这里r0为内,r1为外
r1 = 0.73r
r0 = 0.415r
label_size = 9p
label_font = condensed
# label之间径方向间隔
rpadding = 0r
# label之间角方向间隔
padding = 0r
# 为邻近或者同一位置的多个label设置snuggle显示方式
label_snuggle = yes
# 这里的距离为角方向
max_snuggle_distance = 1r
# 与snuggle排列显示相关的参数,值越大,计算速度越快,但准确性下降
snuggle_sampling = 1
# 值越大,准确性越低
snuggle_tolerance = 0.25r
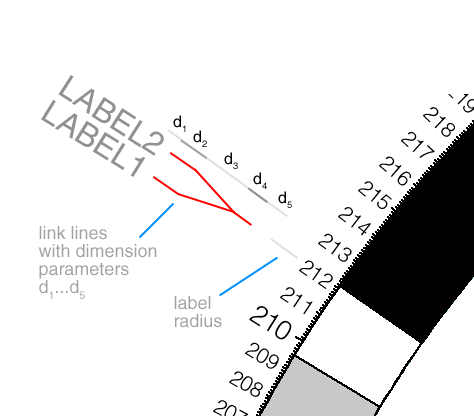
# 这里的link是指引label的连线,具体见下图
show_links = yes
link_dims = 4p,2p,5p,2p,2p
link_thickness = 2p
link_color = grey
<rules>
<rule>
# 为omim相关基因设置颜色
condition = var(value) =~ /omim/
color = blue
# 继续判断后续rule
flow = continue
</rule>
<rule>
# 为cancer相关基因设置颜色
condition = var(value) =~ /cancer/
color = red
label_font = bold
flow = continue
</rule>
<rule>
condition = 1
# 匹配正则表达式
value = eval(replace(var(value),qr/_.*/,""))
</rule>
</rules>
</plot>
</plots>
我们首先看一下关于label的link参数设置:
 最后看一下我们text模块的效果图:
最后看一下我们text模块的效果图:
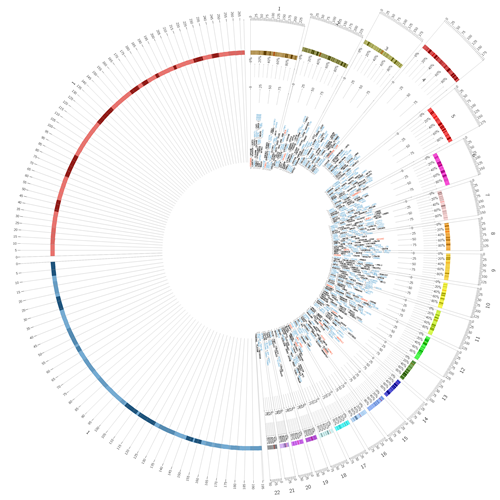 这里有一个小技巧,text不止可以是文字,也可以是符号,如设置
这里有一个小技巧,text不止可以是文字,也可以是符号,如设置label_font = glyph,再将相应数据的value进行更改即可,比如可在上述配置中更改规则:
<rule>
condition = var(value) =~ /cancer/
label_font = glyph
value = N
</rule>

